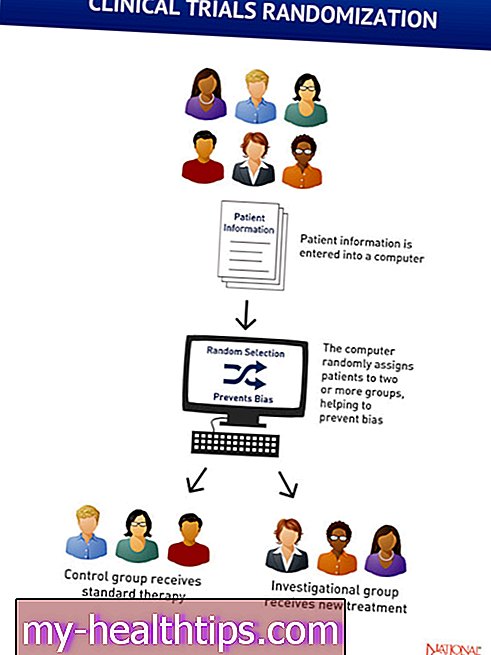
Spinal muskelatrofi (SMA) er en sjælden genetisk sygdom, der gradvist påvirker motorneuroner i rygsøjlen og hjernestammen. Det fører til svaghed hos frivillige muskler, som blandt andet kan påvirke tale, spise, gå og trække vejret.
SMA diagnosticeres typisk ved genetisk testning eller screening af nyfødte hos spædbørn og børn. Som bemærket i en artikel fra 2019 fra American Academy of Pediatrics, er det den mest almindelige arvelige årsag til børnedødelighed.
Der er fire typer SMA: type 1, type 2, type 3 og type 4.
Ifølge Muscular Dystrophy Association har børn, der viser symptomer ved fødslen eller i barndommen, ofte type 1 SMA, hvilket i væsentlig grad påvirker motorfunktionen. Jo tidligere symptomer begynder, jo større er virkningen, idet type 1 er den mest slagkraftige.
Men en ny innovativ genterapi, der blev godkendt i maj 2019 til behandling af børn under 2 år, giver håb for familier, der er ramt af SMA.
Hvad er en-dosis genudskiftningsterapi til spinal muskelatrofi?
Onasemnogene abeparvovec-xioi (mærke navn Zolgensma) er den første genterapi, der er godkendt til behandling af børn, der lever med SMA.
I maj 2019 godkendte Food and Drug Administration (FDA) Zolgensma, en intravenøs enkeltdosisbehandling, der er målrettet mod årsagen til SMA.
Specifikt er Zolgensma indiceret til børn 2 år og yngre uden svaghed i slutstadiet.
Enkeltdosis genterapi gives som en engangsinfusion i venen ved hjælp af en IV. Proceduren tager flere timer, hvor infusionen kører i 60 minutter.
Når infusionen er afsluttet, overvåger sundhedsteamet dit barn i 2 timer. Både under infusionen og derefter overvåges dit barns vitalitet. Opfølgningsaftaler, der inkluderer laboratorietest, kræves i op til 1 år.
Hvordan virker det?
SMA påvirker motoriske nerveceller i rygmarven. Denne sygdom er forårsaget af en arvelig defekt SMN1 gen. Dette får børn til at have problemer med at holde hovedet ope, trække vejret og synke.
Mutationer forårsaget af SMN1 genet klassificeres baseret på alder og sværhedsgrad, hvor den mest almindelige og sværeste er infantil-debut SMA. Desværre overlever mange børn, der er ramt af type 1 SMA, ikke tidligere tidlige barndom.
Den eneste engangsdosis af Zolgensma er målrettet mod den genetiske grundårsag til SMA og erstatter funktionen af den manglende eller ikke-arbejdende SMN1 gen med en ny arbejdskopi af et humant SMN-gen. Dette hjælper motorneuroner med at fungere ordentligt.
Det er vigtigt at bemærke, at Zolgensma ikke ændrer sig eller bliver en del af barnets DNA.
Forskere antyder, at jo tidligere børn får genterapi med spinal muskelatrofi, jo bedre er resultaterne. Børn, der får denne engangs intravenøse administration af Zolgensma, kan bemærke en forbedring i muskelbevægelse og funktion.
Kliniske forsøg viser også et nedsat behov for åndedrætsstøtte og en forbedring i overlevelsesrate. Forsøg har ikke fokuseret på børn med avanceret SMA.
Er det sikkert?
FDA rapporterer, at sikkerheden ved Zolgensma er baseret på både igangværende og afsluttede kliniske forsøg, der studerer i alt 36 pædiatriske patienter med infantil-debut SMA.
Ifølge data er de mest almindelige bivirkninger af Zolgensma forhøjede leverenzymer og opkastning.
Børn med en eksisterende leverinsufficiens har en øget risiko for alvorlig leverskade, hvis de behandles med Zolgensma. På grund af dette skal leverfunktionen vurderes inden behandling og overvåges i mindst 3 måneder efter behandling i henhold til sikkerhedsoplysninger fra Novartis.
Grundig screening og omhyggelig styring af overførsel efter gen er afgørende for sikkerheden og effektiviteten af erstatningsterapi med onasemnogen abeparvovec-xioi.
Resultater
Eksperter er håbefulde om fremtiden med SMA-genterapi.
Ifølge en undersøgelse fra 2020, der blev offentliggjort i tidsskriftet Pediatrics, viste sikkerhed og tidligt resultat fra de første 21 børn (alderen 1 til 23 måneder), der blev behandlet i Ohio, at genoverførsel tolereredes godt hos børn 6 måneder og yngre.
Ældre børn oplevede dog højere stigninger i aspartataminotransferase, alaninaminotransferase og γ-glutamyltranspeptidase, hvilket krævede en højere dosis prednisolon.
Samlet set mener forskere, at resultaterne af undersøgelsen er lovende. Mere specifikt rapporterer de, at symptomatiske patienter oplevede funktionelle forbedringer i motorisk funktion, både subjektiv og objektiv.
Hvad mere er, forskere rapporterer også, at fem børn, der blev behandlet før symptomdebut, ikke udviklede tegn på svaghed, der er typiske for SMA.
En bekymring at bemærke er de høje omkostninger ved lægemidlet. Derudover er Zolgensma hos patienter med avanceret SMA ikke blevet evalueret.
Der er stadig igangværende kliniske forsøg, der tester effektiviteten og sikkerheden af Zolgensma hos forskellige patienter, der repræsenterer forskellige aldre og SMA-typer.
Andre behandlinger for SMA
I betragtning af den nylige godkendelse til brug og høje omkostninger ved Zolgensma er der behov for fortsat forskning. På dette tidspunkt er godkendelse til brug også begrænset til personer under 2 år.
Andre FDA-godkendte behandlinger, der i øjeblikket anvendes til behandling af SMA, inkluderer Spinraza og Evrysdi.
Spinraza er godkendt til alle aldre og typer af SMA. Det er en intratekal injektion, hvilket betyder, at den injiceres i spinalvæsken og skal administreres af en sundhedsperson. Dosering involverer fire injektioner i løbet af de første 2 måneder efterfulgt af vedligeholdelsesdoser hver 4. måned.
Evrysdi er godkendt til dem, der er 2 måneder og ældre. Det er en daglig oral medicin, der tages derhjemme.
Der er andre behandlinger, der stadig undersøges.
Tag væk
Spinal muskelatrofi er en sjælden, men alvorlig genetisk sygdom, der påvirker det perifere nervesystem, centralnervesystemet og frivillig muskelbevægelse. Børn med type 1 SMA har større indflydelse på motorfunktionen.
Resultater fra igangværende kliniske forsøg viser lovende forbedringer i livskvaliteten for dem, der er ramt af SMA, takket være genudskiftningsterapi.
For at få flere oplysninger om en-dosis genudskiftningsterapi til spinal muskelatrofi, skal du tale med din læge.